HASL 4.10 Manual: Chapter Two
A pharmacophore has been defined as a "minimum collection of atoms spatially
disposed in a manner that elicits a biological response." With the advent
of more powerful computationally driven approaches, a number of methods attempt
to define the pharmacophore for a given set of compounds. One such method is
HASL (Hypothetical Active Site Lattice). The basic methodology of the HASL
approach is as follows. The molecular structures of the compounds are converted
into lattices (regularly spaced sets of points). Each point in space is assigned
a fourth variable which describes that type of atom at that point. The lattices
of all molecules are merged to form a composite lattice describing the occupied
space. As will be explained, each lattice point also is assigned a partial
activity value. The summation of the values for any one set of points (i.e., any
one molecule) is equal to the activity for that molecule.
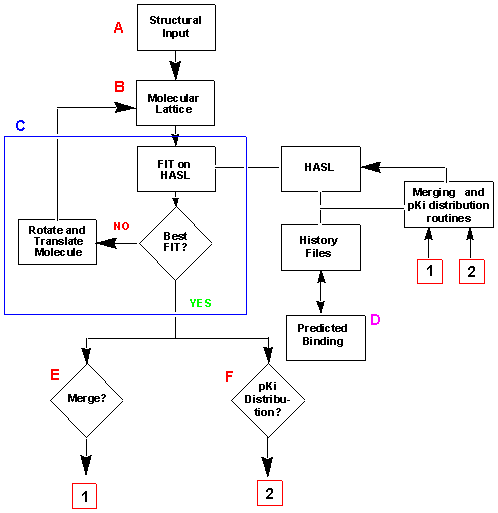
Below is a flow chart outlining the basic processes of the HASL methodology.
The different steps (A-F) and calculations are explained in the text.
A variety of methods are known for the representation of molecules in three
dimensional space, such as steric mapping techniques, molecular volumes, and
molecular shape descriptors. When using HASL, the Cartesian coordinates of an
energy-minimized model are converted to a set of equidistant points arranged
orthogonally to each other, separated by a distance (referred to as the
resolution). All points lie within the van der Waal's radii of the atoms of
the constituent molecules. This framework of points is the molecular lattice,
upon which much of the calculations of this method are based. The number of
points within the lattice is dependent on the molecule size and the chosen
resolution.
As mentioned above, the 4D molecular lattices constructed for each molecule contain
information for four variables: the x, y, and z Cartesian coordinates and a
physiochemical descriptor. For the HASL methodology, a simple indicator variable
was chosen - H, the HASL type. This variable is loosely based on the
quantitative assessment of hydrophobicity derived from a variety of atom types
reported for dihydrofolate reductase inhibitors. The possible values for H
are the integers -1, 0, or +1, which roughly correspond to atoms of low, medium,
or high electron density, respectively. These H values are used to overlay
different structures with an equivalent electronic sense. Thus, similar
atomic characteristics of two molecules can be aligned in three-dimensional
space.
To see a complete listing of the H values for the most common atom types,
please click here.
For each molecule in the data set, there will be an accompanying activity value
for that molecule. Thus, depending on the action of the particular molecule,
such values of activity may include values of Kd, IC50, Ki or others.
For current purposes, we will use -log Ki, of pKi.
The activity, or pKi, of each molecule is associated with the molecular lattice for
each molecule. As a first approximation, the total pKi of the compound is distributed
evenly among its lattice points. For example, if the lattice contains 20 points, then
each point would bear 1/20th of the total activity. Obviously, this is a simple first
approximation. However, because the partial pKi distribution is made without regard
for the internal molecular heterogeneity, this procedure prepares for a separate
redistribution of the activity data for a series of compounds incorporated into the
HASL model.
The comparisons of molecules can be carried out by comparisons of their
respective molecular lattices. First, the generation of a four dimensional (4D)
lattice produces a stationary reference. Next, a second 4D lattice is constructed
and compared to the first, stationary lattice. The degree of matching between
the two molecules is based on the degree of correspondence between the two
lattices. The more points the two lattices have in common, the higher the
match between the two molecules. The degree of matching is quantified by
analysing the FIT,
FIT = L(common)/L(ref) + L(common)/L(molecule)
where FIT is the sum of the fraction of the molecular lattice points and the
fraction of reference lattice points found to be in common. Thus, a perfect
match of two molecular lattices corresponds to a FIT = 2. This procedure
provides a quick means of gauging the progress of molecular matching.
The molecular lattice, as constructed, is a geometric representation of the
space and the nature of that space which is occupied by the
molecule. Biological activities, such as enzyme inhibition data, is then
associated with the lattice so that interactions between a particular molecule
and the eventual HASL can be modeled. In the initial development of the HASL method, enzyme
inhibitory data, as pKi, was used. Of course, any biological or
chemical data can be used.
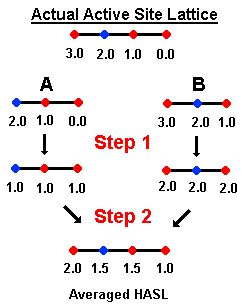
To illustrate the mergining routine, we will consider two molecules,
A and B,
with pKi's of 3.00 and 6.00, respectively, and we will use the
above diagram.
After fitting the 4D lattice of a second molecule to the first, the information
in both lattices is merged. (See Figure #) This results in a composite lattice which
contains all points present in either initial lattice. This composite
lattice is represented by the four linear points, directly under
"Actual Active Site Lattice." The different colors represent characteristics
(e.g., elctron density) of the molecular lattice. Molecule A occupies the
three right points (blue, red, red) of the lattice, whereas
molecule B occupies the three left points (red, blue, red) of the lattice.
The two molecules thus have the two middle points in common.
In the first step (Step 1), partial pKi distribution
is averaged. For Molecule A (pki = 3.0), one unit of pKi is distributed per point. In the next step (Step 2), the partial
pKi is averaged at common points. For the above example, the two middle points
are averaged yielding 1.5 at the middle red and blue points.
This averaging step produces an Averaged HASL. In the average HASL, the
left most point corresponds to a point in space occupied by Molecule B alone; the
right most point to Molecule A alone.
Of course, this
simple averaging does not provide a solution to the problem. From the averaged
HASL, the predicted activity of Molecule A is 4.0 (sum of points occupied by A 1.5+1.5+1.0) and for Molecule B is 5.0 (sum of points occupied by B).
The averaged HASL
is then used as a starting point for additional refinement as illustrated
in the above scheme. For example,
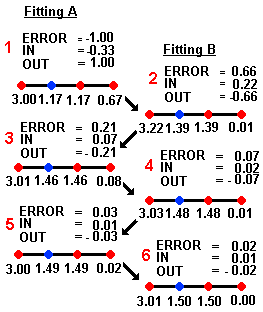
molecule A (its molecular lattice, to be precise) is fitted to the averaged
HASL and this yields a set of corrections referred to as IN
and OUT whose
errors are dependent on the overall error in predicted activity (referred to
as ERROR).
IN = (correction calculated as ERROR)/NI
where NI = number of lattice points in the overlap.
OUT = (correction calculated as -ERROR)/NO
where NO = number of lattice points outside the overlap.
These corrections are determined as shown in the above scheme and are then applied to
the current partial pKi values assigned to each HASL point. IN corrections
are applied to only those points that the particular molecule (A) and the HASL
have in common; OUT corrections are made to those points which the molecule and
the HASL do not share.
One merging cycle is complete when the procedure is repeated with every molecule
what was used to create the HASL and predictivity checked. For the present
example, an iterative cycle would be the fitting of A, followed by appropriate
corrections, and the fitting of molecule B, followed by the appropriate
corrections. This iterative cycle is repeated until an acceptable error of
predictivity is reached.
The initial HASL that emerges from the Merging Routine usually contains a large
number of lattice points compared to the number of data points (i.e., the number
of molecule). Thus, it would be prudent to reduce the model to a smaller, more
robust subset of points which retains the predictiveness while minimizing the
possible overfit of the data. To do obtain this goal, a process of HASL
trimming is performed.
As the number of points are reduced in order to locate the most significant lattice
points, the process must ensure the following:
(1) The initial model has incorporated all potentially relevant points, and
(2) the process itself does not remove the relevant points.
From the above, it appears that building a three-dimensional pharmacophore from a HASL requires
an initial, detailed model, with small lattice spacing, to ensure that all or most atoms
in each molecule are represented. Also, to prevent the loss of relevant points in the trimming
process, the effects of different trimming methods on the resultant models' predicitivities can
be examined. If the model retains good predicitivity, then we can assume that the model has
retained the important lattice points.
The actual trimming process contains two steps that are performed in an iterative fashion until
an optimal HASL-derived pharmacophore is obtained.
Trimming Process
STEP 1: Removal of those HASL points that currently represent the least significant partial pKi
values (e.g., 10% of all HASL points in the current model that have partial pKi values nearest
to zero).
STEP 2: Iterative distribution of the partila pKi values among the remaining lattice
points to achieve the best correspondence between actual and predeicted pKi.
Note:
This section on the theory of HASL has been adapted from the following two
references:
1) Doweyko, A.M. The Hypothetical Active Site Lattice. An Approach to Modelling
Active Sites from Data on Inhobitor Molecules. J. Med. Chem. 1988
, 31, 1396-1406.
2) Doweyko, A.M. Three-Dimensional Pharmacophores from Binding Data. J. Med.
Chem. 1994, 37, 1769-1778.