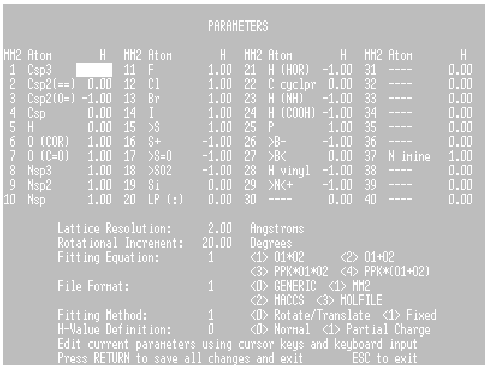
Figure 2. The Parameter Editing Screen.
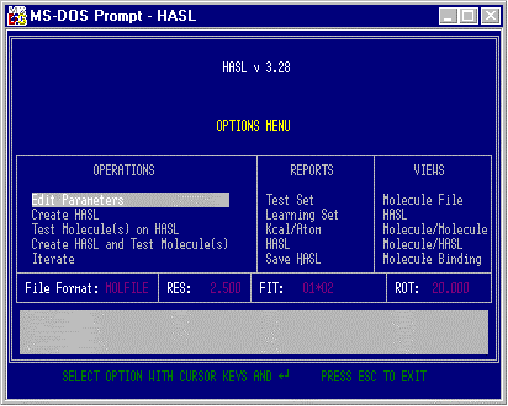
After installation of the HASL files, entering the command “HASL” starts up the user interface program, HASL.EXE and displays the Options Menu screen pictured in Figure 1. It is through the Options Menu that the user identifies the Cartesian file type, the file names, activities, type of fitting paradigm to enact, and what, if any, test molecule files will also be used. The Options Menu also provides a modest set of report choices as well as graphic viewing choices enabling the user to “see” the inter-relationship between model and molecular structure.
The cursor/RETURN sequence is used to select any option on the screen. Pressing escape (ESC) generally aborts the selection and permits the user to choose another. Comments appearing in the Message Area provide further options or indicate the status of ongoing processes.
Note that the Options Menu screen is segmented into several areas: Operations, Reports
and Views. In addition, several key operational assumptions currently selected are
displayed: File Format, RES (resolution), FIT (fitting paradigm), and ROT (rotational
increments in degrees).
Operations
Edit Parameters
Figure 2. The Parameter Editing Screen.
As noted earlier, since most operational choices are stored in the parameter file, PARAMS.DAT, the user should begin the modeling session by selecting “Edit Parameters” (positioning the highlighted cursor bar over this option and pressing RETURN).
The following options will appear in the Message Area:
(E)dit existing parameter fileIf the user is interested in making minor changes in the file, pressing “E” will bring up the file as it is currently defined. Choosing “C” will substitute the previous parameter file with the default version of PARAMS.DAT (the one that the program creates automatically if no other is present.
(C)reate new parameter file or
ESC to cancel this option
The Parameters screen is shown in Figure 2. Editing of parameter values is accomplished by placing the cursor over the current value and entering the desired value. Entering values outside acceptable limits will elicit an error alert and the user will be prompted to re-enter an acceptable value. When all changes have been made, pressing RETURN will bring up an option to save all current selections in PARAMS.DAT or under a user-selected file name (This user- selected file name cannot be accessed by HASL unless the user renames the file “PARAMS.DAT” at an operating system level). Pressing ESC will abort the procedure, retain previous parameter selections, and return the user to the Options Menu.
Parameters 1-40 Atom Types
The first 40 parameters are atom type definitions based on MM2 (Allinger) atom
types. Regardless of the type of Cartesian coordinate files used in modeling,
HASL uses the MM2 atom types as the basis of its H-value assignments. Thus,
for Alchemy *.MOL files, HASL will automatically convert the Alchemy atom
types defined in the *.MOL files to H-values according to the definitions listed in
the PARAMS.DAT file. The default set of H-values represent a simplification of
atom type definitions wherein only three atom types are considered significant:
H=1, atoms rich with electrons, e.g., O, N, S, Cl
H=-1, atoms poor in electrons, e.g., C as in C=O, H as in OH
H=0, atoms neutral in electrons, e.g., C or H as in CH3
The H-values are used by HASL to assign atom type character to each lattice point derived from a molecule. These values are used as integers within the program. The user is free to experiment with other H-value assignments; for example, the user can opt to consider only the shape of a molecule by assigning the same H-value to all atom types (e.g., H=0). It is also possible to have H- values automatically assigned based on partial charge information (see parameter 46).
Parameter 41 Lattice Resolution
Lattice resolution can be set to any Angstrom value and represents the point spacing in the HASL model. The choice is somewhat critical: if the resolution value is set too high, the resulting lattice of points may be spread too wide apart to capture essential molecular details; and if it is too small, the lattice will be composed of too many points and suffer possible over-fitting of subsequently incorporated binding data. In its current state of development, based on the work cited in the references (see Theory), the optimum choice for resolution appears to be in the range of 2-3 Angstroms. It is at about 2.5 Angtroms that most atoms >95% in most organic molecules are represented by an equally-spaced three- dimensional grid of points.
Parameter 42 Rotational Increment
Parameter 43 Fitting Equation
Parameters 42 and 43 apply only if the fitting method (parameter 45) is selected to rotate/translate molecules during their alignment and superpositioning. The fitting procedure first translates each new molecule along each major axis (x,y,z) one resolution unit, finds a best fit, then rotates the molecule along each axis using the rotational increment (parameter 42), finding a best fit, and repeats the process until a maximum fit is achieved between the molecule and its target lattice of points. The proper choice of rotational increment depends on the computing power at the user’s disposal, since a small value will elicit a very large number of calculations. Typically, at the PC level, a choice of ca. 20 degrees appears to represent a reasonable compromise between speed and efficiency. The degree of fit is determined by the overlap between the molecular lattice of points and the target lattice (the current HASL model), using the equation selected in parameter 43. The equations refer to O1 and O2, the fraction of HASL points shared with a molecular lattice and the fraction of molecular lattice points shared with the HASL, respectively. In equations 3 and 4, the partial pKi (assumed binding value) is given as PPK and can be used to steer the incoming molecule toward the HASL by weighting fitting in terms of binding potential as well as points in common. Although the experimentalist is free to choose between fitting paradigms, it is recommended that fitting be limited to a simple overlap paradigm (equation 1 or 2) to avoid the potential for “drift” as new molecules are incorporated into a growing HASL model.
Parameter 44 File Format
There are four Cartesian coordinate file types directly supported, i.e., not requiring conversion outside the program. These are:
1. Generic - These files are of the format that HASL uses internally. This same format is used in IMAGE.DAT and *.HSL files.
2. MM2 - Allinger MM2 file format.
3. MACCS - Molecular Design, Limited (MDL) file format.
4. MOLFILE - Alchemy (Tripos) file format.
The format definitions of files created by other commercial applications, as found in this manual, are to be considered as approximations made by this author. Further descriptions of these file architectures are detailed in Appendix B.
Parameter 45 Fitting Method
The fitting method has been described to some degree in the discussions of parameters 42 and 43. In choosing fitting method 0, the HASL model will be developed by rotating/translating each new molecule, seeking out a best fit, before it is incorporated into the current HASL. Fitting method 1 uses the Cartesian coordinates as they exist in each Cartesian coordinate file, i.e., the molecules are assumed to have been optimally pre-aligned or superposed.
Note to purchasers of HASL source code: The incorporation of module H13.FOR will allow an experimental type of fitting to occur when parameter 45 is set to 2. This module will pre-position the new molecule in such a way as to minimize the differences in H-value orientation, i.e., incoming molecules will be aligned by orienting their H-value distribution in a manner most similar to the H- value distribution already present in the HASL. This module is strictly experimental and would appear to suffer the same pitfalls that the use of fitting equations 3 and 4 engender, i.e., a tendency for the model to grow larger-sized as new molecules are incorporated (drift).
Further Note: Although H13.FOR has been incorporated into the executable PC version, parameter 45 cannot be set to 2 within HASL. Since this module was and is considered experimental and unreliable, its implementation has not been made generally available. The adventurous user can circumvent this block by using a text editor to modify line 45 of PARAMS.DAT to “2”.
Parameter 46 H-Value Definition
Normal (0) H-value definitions are those listed as parameters 1-40. The selection of the partial charge option is only viable if the Cartesian coordinate file contains partial atomic charges. This option is available if the file type is MOLFILE (see Appendix B). The resulting H-value obtained in this manner is an integer computed using the following equation: H-value = INT(10*Partial Charge).
Create HASL
The creation of a HASL model is enacted after the user completes making parameter selections. Upon selecting “Create HASL,” the user is prompted to choose between creating a new HASL or adding molecules to a pre-existing HASL. Please note that the creation of a new HASL will erase pre-existing information in HISTORY and MOL*.FIT files, and obliterate the RECEPTOR file containing the old HASL model. If the user intends to save the old HASL model before starting a new one, it is necessary to enact the “Save HASL” option (see Reports).
If the “Add to HASL” option is selected, the user can then decide whether to add new information to the current HASL or to a previously saved model. To retrieve a saved model (including all pertinent files) the user enters the directory name under which it was saved.
The program requests the Cartesian coordinate file name and activity for each molecule either through direct keyboard entry of each or by identifying an ASCII file which contains these filenames and activities (each line in the ASCII file consists of a Cartesian coordinate file name and activity value separated by a space; this file can be created using a text editor or spreadsheet).
In the keyboard entry mode, upon entering a file name (with subdirectory prefix, if necessary), the program immediately checks that subdirectory to see if the file exists. If the file is not found, then the user is so informed and is asked to re-enter the file name, or press “?” to shell out to DOS. Shelling out to DOS permits the user to check the file name and directory settings, after which, entering “EXIT” will return the user to the file name prompt.
The nature of the activity value plays a significant role in developing a viable model. In HASL modeling, the assumption is that the activity value is linearly proportional to the free energy of binding. The use of pKi or -log(Ki), where Ki is an enzyme/inhibitor dissociation constant, is an example of such an activity scale, since pKi is related to the free energy of binding by the relationship delta G = -RTlnK (discounting significant contributions from entropic changes resulting from binding). Despite this apparent limitation, HASL methodology has been successfully applied utilizing a variety of in vivo data. The choice of the activity value is up to the experimentalist, keeping in mind that the eventual biological effect being monitored may be significantly influenced by factors other than pure binding at a receptor site, e.g., transport, and metabolism.
If more than one molecule is used to create the HASL, then after all file names and activities are entered, the user is prompted to enter the number of times to iterate the model. This operation refers to the procedure by which the program, namely ITERATE.EXE, redistributes partial binding values among available lattice points in such a way as to minimize the error in predicting activities of the learning set. Any number of iterations can be entered and the progress of the iterative process is dependent upon the number of molecules and the nature of their activity values, as well as the target error limit entered following the subsequent prompt. Typically, for 10-50 molecules with well-defined pKi values, about 100-300 iterations can bring the predicted error down to 0.001. There is no harm in selecting an iteration number too small, since the process can be repeated using the “Iterate” option in Operations. During the iteration process the receptor description yielding the smallest error in prediction is saved to disk.
The resulting HASL model is saved in a file called RECEPTOR and the building process is recorded in a file called HISTORY.
After all prompts have been addressed, the program creates MOL*.FIT files and a batch file, HSERIES.BAT, which is launched under DOS. The batch file contains a series of commands which make calls to various HASL executable modules and lists of file names/activities. When these operations at the DOS level are complete, the user is returned to the Options Menu. The nature of the batch file for this and other operations is discussed further in Appendix A.
Test Molecules The election of the “Test Molecule(s) on HASL” option assumes the existence of a previously created HASL model. This option is primarily used to predict the activities of a set of as yet untested molecules. The number of molecules, file names and activities are entered as described for the creation of a HASL. Although the program prompts the user to enter activity values for the test molecules, it is naturally possible that these values do not exist, in which case, entering any numerical value, including zero, will suffice without affecting the program, since these values will only be used for comparative purposes. The user will be further asked to either “Erase” or “Append” the HISTORY file. Electing to “Erase” will start a new HISTORY file with the first test molecule data. Electing to “Append” will add the new data to the previously-generated HISTORY file. The test molecule report will include previously-fitted molecules if “Append” is elected (see Reports). The program creates MOL*.TST files and a batch file, TSERIES.BAT, which is launched under DOS. Upon completion of the run, the user is once again returned to the Options Menu.
Create HASL and Test Molecule(s)
The use of this option simply provides the user with the ability to set up both the HASL creation process and its testing automatically. The program utilizes the prompts already discussed to create both MOL*.FIT and MOL*.TST files (containing file names and activities), and a master batch file, SERIES.BAT, which contains the necessary elements of HSERIES.BAT and TSERIES.BAT. Upon entering all the required data, the program launches SERIES.BAT and returns the user to the Options Menu upon completion of both operations.
Reports
Report options provide the user with a convenient way to view summaries of results which can be directed to the screen, printer, or file.
Test Set
The fitting results and activity predictions for the most recently run test set of molecules are listed in this report. Note that this information is obtained from the data residing in the current HISTORY file (which may contain data from previously fitted molecules if the “Append” option was elected). An example of this report is shown in Figure 3.
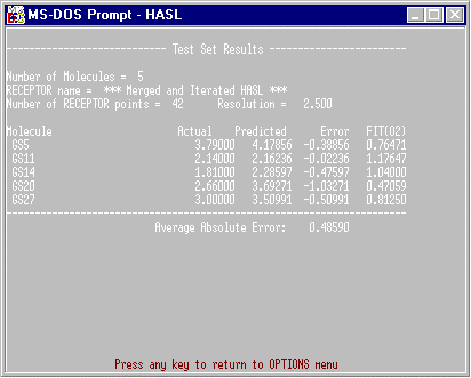
Figure 3. Example Test Set Report.
Learning Set
In the learning set report, the molecules used in the most recent HASL creation are listed along with a comparison of actual to predicted activities based on the information contained in ITERAT.DAT. An example of this report is found in Figure 4.
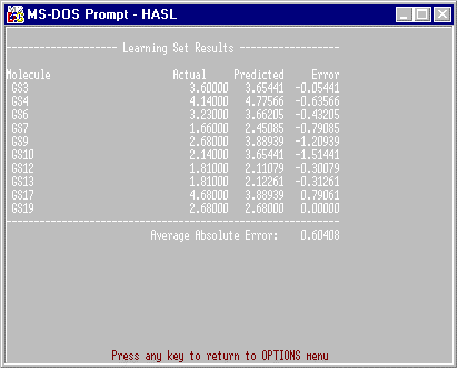
Figure 4. Example Learning Set Report.
Kcal/Atom
The kcal/atom report provides an atom-by-atom estimate of the free energy of binding for the last-fitted molecule. The estimates are based upon the assumption that activity values are in the form pKi and that no significant entropic contribution occurs during binding. The estimates are built up by summing the partial binding values coincident with the molecule’s overlapped lattice of points. Since each atom can be related to a HASL point carrying some partial binding value, the sum of partial binding values corresponding to that atom’s presence is used to calculate the free energy of binding contribution by that atom. This is done by using the relationship: delta G = 2.303 RT Sigma(partial pKi).
Of course, if activity values other than pKi are used, no such direct estimate is possible, however, relative importance of atoms in the molecule may still be highlighted by this procedure. An example of a partially-listed kcal/atom report is shown in Figure 5.
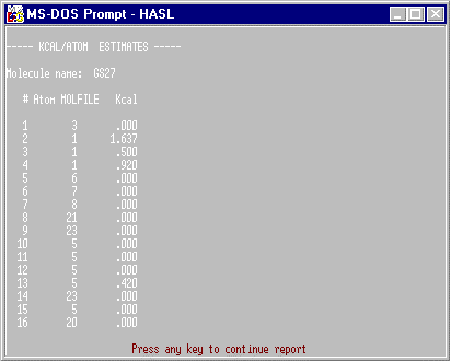
Figure 5. Example Kcal/Atom Report.
HASL
This report lists the contents of the file RECEPTOR which represents the essential elements of the HASL model: the name of the model (which is a default name generated by the program), the resolution used to create the model, and the number of lattice points making up the model. In addition, each lattice point is listed (x,y,z) along with the H-value assigned to that point, the partial pKi (HASL always assumes the value to be pKi), and the number of molecules that was responsible for the existence of that point, i.e., the number of molecular lattices sharing that point. A partial listing of such a HASL model description is found in Figure 6.
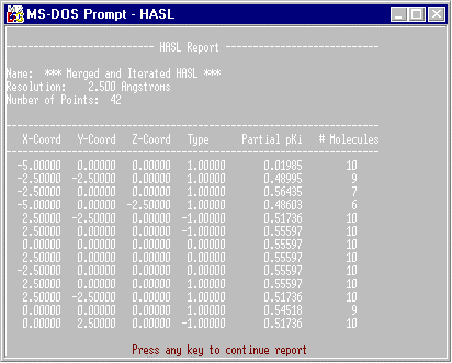
Figure 6. Example HASL Report.
Save HASL
This option allows the user to save a previously-generated HASL model to a directory from which it can be later retrieved. The user is prompted for a directory name after which the program enters DOS, creates the directory (if not already present), and copies all relevant files to that directory.
The NAMELIST.HSL file contains a list of all MOL*.FIT file names used in HASL model creation. The actual Cartesian coordinate files ARE NOT copied. Thus, the user should insure that these coordinate files are not moved or deleted from their directory.
When a saved HASL is retrieved under the “Create HASL” option, all MOL*.* files are deleted from the working directory prior to the retrieval process.
Views
The graphics capabilities supported by HASL are limited to EGA/VGA PC screens. Images are shown in four color displays. No graphics screen printing is supported directly, however the user may opt to perform a graphics screen dump using appropriate system software.
Molecule files, the HASL model, molecule/molecule and molecule/HASL overlays are graphically accessible. Note that the “molecule/molecule” option seeks out *.HSL files (previously created to store molecular alignment as “fitted” to the HASL). The default selection (hitting the RETURN key twice) under this option automatically displays the MOLMOL file (the first molecule used in the creation of the HASL and the last molecule fitted to the HASL). In addition, the putative binding of the last-fitted molecule can be studied graphically using the “Molecule Binding” option (discussed later). When a specific view is selected from the Options Menu, the selected file(s) is converted to a set of coordinates in file IMAGE.DAT and automatically accessed by the module HDRAW.EXE. Once presented on the screen, the image can be manipulated in a number of ways by pressing the highlighted letter, halting the action with the SPACE bar, and pressing ESC to recolor the image:
U.......... Up
D.......... Down
L........... Left
R........... Right
S........... Shrink
E.......... Enlarge
X, followed by # of degrees .......... rotates about the X-axis, # degrees at a time
Y, followed by # of degrees .......... rotates about the Y-axis, # degrees at a time
Z, followed by # of degrees .......... rotates about the Z-axis, # degrees at a time
RETURN........... returns the user to the Options Menu
Pressing F1 will present these options in the form of a HELP Screen.
In addition to the viewing options discussed thus far, there is an “undocumented” graphics capability available to the user. If the working directory also contains the program ACROSPIN (Acrobits, P.O. Box 26871, Salt Lake City, UT 84126-0871), pressing F10 while in the Options Menu screen will launch the creation of a file called "IMAGE," which is accessed by ACROSPIN and provides the user with extremely fast graphics viewing capabilities of the last-selected View. This particular capability may be incorporated as object code in the next version of HASL for Windows95.
Molecule Binding
Selecting this view first provides the user with a bar chart synopsis of the estimated kcal/atom values distributed in the last-fitted molecule (Figure 7). For example, 7% of the atoms in that molecule have estimated kcal (binding) values between 0.88 and 1.26. By selecting a “lower cutoff value for the red color” the atoms in the molecule with binding values greater than that entered will be colored red. The next prompt asks a similar question
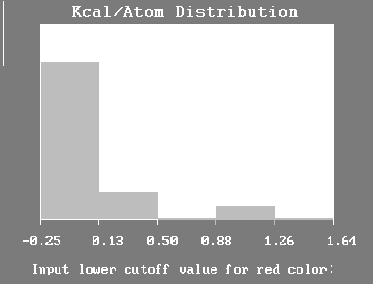
Figure 7. Molecule Binding - Highlight Selection Screen.
for the color yellow. In this way the user can selectively highlight those features in the molecule that HASL has determined as most or least significant to binding. The distribution observed in this analysis is based on that obtained by fitting a molecule to the current HASL and using the kcal/atom estimates found in file KCAL.DAT.