CHAPTER 6
Input File Formats
The Molconn-Z software package has provision for several formats of files for the input of molecule structure. The
flow of information from input structure files to output is described in
Chapter 4.
The input molecular structure information is contained in the .B file when SMILES code or the standard
MOLCONN format are used. When other formats are used, the connection table information is stored in separate
molecule files, each containing a single connection table; then the .B file is simply a list of those file names,
as described below. The MDL structure data file format (SDFile) includes the actual Molfiles and lines of data.
There are a variety of options for structure input format, including the standard MOLCONN format for connection tables, as follows:
- Standard MOLCONN file format
- SMILES code
- ChemDesign CSSR files
- ChemLab or MicroChem file format (no longer supported)
- ChemDraw file format
- MDL Informations Systems, Inc. Molfile format
- MDL Informations Systems, Inc. SDFile format
- SMILES using Daylight Toolkit (unix versions - optional)
- User designed file format (by contract with Hall Associates only).
The .B file can be created or obtained in several ways:
- using an editor and entering directly the necessary information as described below for SMILES code or standard MOLCONN form;
- using a graphic type input or database which also produces a connection table which corresponds to one of the formats described below.
- obtaining the connection table from a preexisting database and converting it to the format described below.
Input using standard MOLCONN format is illustrated in detail in the next section. Use of the other formats
is described in subsequent sections. However, no attempt is made here to give a description of the
particular format. Rather, our purpose is to illustrate how such formats may be utilized in the Molconn-Z
software. For specific information about each of the formats, the user is directed to the appropriate
company representative or literature source.
- MOLCONN .B FILE Format
The input file which contains molecule descriptions is called the .B file, which stands for BOND
file. Essentially, the .B file contains connection tables in the form described below.
The input .B file in Standard MOLCONN Format contains the connection table for each of the molecules in the
data set. The information for each molecule consists of three parts:
- ID line,
- Atom Identifiers and Connections,
- Molecule Terminator.
- ID line:
a) Molecule ID : the user's sequence number (1 to 9999) for the molecule which can be used for
retrieval of computed information.
b) Molecule name : the user's name for the molecule in whatever form the user wishes; 60 character
limit on the name length.
- Atom Identities and Connections
There is one line for each skeletal atom: Atom ID, NH, Atomic Symbol, ID s of all bonded atoms, VALD.
a) atom ID : The sequence number of the atom in the molecular skeleton. Numbering is arbitrary but
it may well be useful to conform to some standard or consistent numbering scheme.
It should be noted that the maximum number of atoms permitted by Molconn-Z is limited
by memory size and the limits put into PARAM.DAT at compilation time.
b) NH : the number of hydrogen atoms bonded to the skeletal atom; e.g., 2 in -CH2-, 1 in -OH, 0 in Cl,
2 in -NH2.
c) Atomic Symbol : standard atomic symbol for the atom. Recognized symbols include atoms of atomic
numbers 1 - 55. Chi indexes have been tested for elements B, C, N, O, F, Si, P, S, Cl, Br, I; Kappa
alpha values are established for H, B, C, N, O, F, Al, Si, P, S, Cl, Ga, Ge, As, Se, Br, Sn, Sb, Te, I.
Special Cases:
H may be used for a special role as in hydrogen bonding.
Q may be used for any atom not treated by Molconn-Z. User must
supply valence delta value.
d) IDs of all bonded atoms : the IDs of all the skeletal atoms bonded to the atom.
e) VALD (OPTIONAL) : an alternate value for dv may be supplied here by the user; used when no
standard value is available in Molconn-Z. The user supplied value must contain
a decimal point and is limited to a maximum of five decimal places, e.g.: 3,0,Se,2,4,0.22222
The general nonempirical relation may be a guide: dv = (Zv - h)/(Z - Zv -1).
- Molecule Termination
Each molecule is terminated with a '-1' on a separate line.
NOTE: Each quantity described above is separated from the next by either a comma or
a blank (field delimiter). The user can specify either of the field delimiters in the OPTION section
of the program by referring to Option 2 in the first submenu. The standard option is a comma.
- File Termination
The signal to the Molconn-Z program that the end of the .B file has been reached is a '-1'. In general,
the .B file will contain two consecutive lines at the end with a '-1'; one for the end of the last
molecule and one for the end-of-file signal. See the example .B file, SAMPLE.B.
The definitions given above are illustrated by examples below. Further, the file SAMPLE.B (supplied
with the Molconn-Z software) also contains many more examples. See the listing of this file at the
end of this chapter.
Example 1: Serotonin
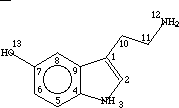
2, Serotonin
1,0,C,2,9,10
2,1,C,1,3
3,1,N,2,4
4,0,C,3,5,9
5,1,C,4,6
6,1,C,5,7
7,0,C,6,8,13
8,1,C,7,9
9,0,C,1,4,8
10,2,C,1,11
11,2,C,10,12
12,2,N,11
13,1,O,7
-1
Example 2: 4-Chlorophenol
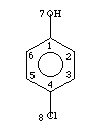
1, 4-Chlorophenol
1,0,C,2,6,7
2,1,1,3
3,1,C,2,4
4,0,C,3,5,8
5,1,C,4,6
6,1,C,1,5
7,1,O,1
8,0,Cl,4
-1
Example 3: 2,2-Dimethyl-5-chloro-pentane-4-one
3, 2,2-Dimethyl-5-chloro-pentane-4-one
1,3,C,2
2,0,C,1,3,7,8
3,2,C,2,4
4,0,C,3,5,9
5,2,C,4,6
6,0,Cl,5
7,3,C,2
8,3,C,2
9,0,O,4
-1
Use of SMILES Code in Input Format
Molconn-Z has several options for use of input file format. The input option is selected as item 1
in the MENU section of the program. To use the SMILES code, the user selects 'S' from the Main
Menu in option 1.
SMILES code was developed by David Weininger (D. Weininger, J. Chem. Inf. Comput. Sci., 28, 31-36,
1988) to provide a string code for the input of molecular structure. The user is referred to this
reference and subsequent papers for the description of the SMILES code and techniques for creation of
SMILES code for molecular structures. The following two structures illustrate the application of
SMILES code. Essentially, the chemical graph is reduced to a tree (noncyclic) graph by removing one
bond for each ring; the atoms between which the bond was broken are labeled with a number. Branches
are enclosed in parentheses.
Examples
The following is the contents of the example input file which illustrates SMILES code,
SMILES.B, from the Molconn-Z software package.
The File SMILES.B as Supplied in Molconn-Z Software:
1, 6-Hydroxy-1,4-hexadiene
C=CCC=CCO
2, Triethylamine
CCN(CC)CC
3, Isobutyric Acid
CC(C)C(=O)O
4, 3-Propyl-4-isopropyl-1-heptene
C=CC(CCC)C(C(C)C)CCC
5, Benzene
c1ccccc1
6, 3-Bromo,methycyclohex-1-ene
CC1=CC(Br)CCC1
7, Cubane
C12C3C4C1C5C4C3C25
8, Tetramethyl silane
C[Si](C)(C)C
9, Morphine
O1C2C(O)C=CC3C2(C4)c5c1c(O)ccc5CC3N(C)C4
-1
Note that the '-1' file terminator is used as the last entry in the file.
Three extensions have been made to SMILES code interpretation for Molconn-Z version 3.0+.
- Ring closure numbers have been extended beyond the range 0 to 9. For higher numbers, a % sign
must be included. For example, for cyclobutane, if one wished, the following string can be used:
C%11CCC%11. This additional feature permits more than 10 rings per structure.
- Ring closure numbers may now be reused within the same string. The SMILES interpreter will,
of course, assume that the second occurrence of a given digit must be paired with the first
occurrence of that digit. For example, biphenyl could be written as follows: c1ccccc1c2ccccc2
or as c1ccccc1c1ccccc1. The string c1ccccc2c1ccccc2 would correspond to an entirely different structure.
- The length of the SMILES string allowed for input has been increased to 256.
Molecule Files from CHEM-X of Chemical Design Ltd.
Molconn-Z has several options for use of input file format. The input option is selected as item 1 in
the MENU section of the program. To use files in the CHEM-X format, the user selects 'X' from the
Main Menu in option 1.
The use of molecule files produced by Chem-X is very easily done. The user first produces the
desired molecule files by using Chem-X in its usual manner. Each molecule file is produced with a
unique name, usually closely associated with the name of the molecule. These molecule file names
are entered into the .B file rather than the connection table information required by the standard
Molconn-Z format. The molecule file names become pointers to the actual connection table information
in the Chem-X molecule files. It is most helpful to have all the Chem-X molecule files in the
directory in which the user is working.
For example, suppose the user has produced Chem-X files for the following molecules: acetic acid and
benzoic acid with the following file names: ACETICAC.CSS, CLBENZCA.CSS (Note: VAX style notation is
used here and the suffix CSSR (shortened to CSS here) is simply an acronym for Cambridge Structure
Search Routine and not necessary for general use.) Then, the Molconn-Z .B file, let's call it CHX.B,
will have the following form:
The File CHX.B as Supplied in Molconn-Z Software:
1, ACETICAC.CSS
2, CLBENZCA.CSS
3, ETHANOL.CSS
-1
It is of the utmost importance that the file names in the .B file be exactly as they
appear in the directory listing. The usual '-1' file terminator is used.
The test input file CHX.B is supplied with the Molconn-Z software along with the appropriate Chem-X
molecule files ACETICAC.CSS, CLBENZCA.CSS, and ETHANOL.CSS.
Chem-X is developed and distributed by
Chemical Design, Ltd.
Oxford, England
Molecule Files from ChemLab and MicroChem(TM)
THIS FILE FORMAT IS NO LONGER SUPPORTED BY HALL ASSOCIATES. IF A USER DESIRES THIS FORMAT, CONTACT
HALL ASSOCIATES FOR A CUSTOMIZED VERSION OF MOLCONN-Z.
MicroChem(TM) is a trademark of Intersoft, Inc.
282 East Woodland Rd.
Lake Forest, IL 60045
Molecule Files from ChemDraw File Formats
Molconn-Z has several options for use of input file format. The input option is selected as item 1 in
the MENU section of the program. For use of files from ChemDraw, select 'D' in option 1 in the Main
Menu.
The use of molecule files produced by ChemDraw is very easily done. The user first produces the desired
molecule files by using ChemDraw in its usual manner. Each molecule file is produced with a unique name,
usually closely associated with the name of the molecule. These molecule file names are entered into
the .B file rather than the connection table information required by the standard Molconn-Z format.
The molecule file names become pointers to the actual connection table information in the ChemDraw
molecule files. It is most helpful to have all the ChemDraw molecule files in the directory in which
the user is working.
For example, suppose the user has produced ChemDraw files for the following molecules : pyridine and
propanoic acid, with the following file names: PYRIDINE.TBL and PROP_ACD.TBL. (Note: VAX style
notation is used here.) Then, the Molconn-Z .B file, let's call it CDRAW.B, will have the following
form:
The File CDRAW.B as Supplied in Molconn-Z Software:
1, PYRIDINE.TBL
2, PROP_ACD.TBL
3, PYRIDIN0.TBL
4, 4CLBIPHE.TBL
-1
It is of the utmost importance that the file names in the .B file be exactly as they
appear in the directory listing. The usual '-1' file terminator is used.
The test input file CDRAW.B is supplied with the Molconn-Z software along with the ChemDraw molecule
files PYRIDINE.TBL , PROP_ACD.TBL, PYRIDIN0.TBL , and 4CLBIPHE.TBL.
ChemDraw is a trademark of Cambridge Scientific Computing, Inc.
875 Massachusetts Ave.
Cambridge, MA 02139
Molecule Files from MDL Information Systems, Inc. Molfiles
Molconn-Z has several options for use of input file format. The input option is selected as item 1 in
the MENU section of the program. The user selects 'M' for use of files in the Molfile format from
MDL Information Systems, Inc. in option 1 in the Main Menu. For a complete description of the Molfile,
see A. Dalby, J. G. Nourse, et al., J. Chem. Inf. Comput. Sci., 32, 244-255 (1992).
The use of molecule files in the Molfile format produced by MDL software is easily done. The user first
produces the desired molecule files by using MDL software in its usual manner. Each molecule file
is produced with a unique name, usually closely associated with the name of the molecule. These
molecule file names are entered into the .B file rather than the connection table information required
by the standard Molconn-Z format. The molecule file names become pointers to the actual connection
table information in the Molfile. It is most helpful to have all the Molfiles
in the directory in which the user is working.,p>
For example, suppose the user has produced Molfiles with the following file names: PHENOL.MOL,
CLPHENOL.MOL, CNPHENOL.MOL, NNPHENOL.MOL, and SNPHENOL.MOL Then, the Molconn-Z .B file, let's call
it MDL.B, will have the following form:
The File MDL.B as Supplied in Molconn-Z Software:
1, PHENOL.MOL
2, 4CLPHENOL.MOL
3, CNPHENOL.MOL
4, NNPHENOL.MOL
5, SNPHENOL.MOL
-1
It is of the utmost importance that the file names in the .B file be exactly as
they appear in the directory listing. The usual '-1' file terminator is used.
The test input file MDL.B is supplied with the Molconn-Z software along with the MDL molecule files
PHENOL.MOL and 4CLPHENOL.MOL.
MOL file format is licensed by MDL Information Systems, Inc.
San Leandro, CA
SDFiles from MDL Information Systems, Inc., using Molfiles
MDL also supports an additional type of file format which includes structure
data in the form of the Molfile. This SDFile also includes provision for an unspecified number of
records which contain data of various types for each molecule. The data may be numerical or
alphabetic. Select 'F' in Menu option 1 for SDFile input format.
This Structure Data file (SDFile) is carefully described in A. Dalby, J. G. Nourse, et al., J.
Chem. Inf. Comput. Sci., 32, 244-255 (1992).
The use of the SDFile format produced by MDL software is easily done. The user first produces the
desired molecule files by using MDL software in its usual manner. These molecule files are
incorporated into the .B file along with the data lines desired by the used, following each
Molfile. The record separating the Molfile from the data records contains 'M END'. See the
example below and the reference given above. The information for each molecule is terminated
by a blank record followed by a record containing $$$$. The whole SDFile is terminated with
a blank record.
For example, suppose the user has produced an SDFile for the following molecules: phenol and
4-chloro-2-nitrophenol. Let's call it SDF.B. It will have the following form:
The File SDF.B as Supplied in Molconn-Z Software:
PHENOL
JFMACCS 8302248414282D 1 0.00213 0.00000 0 JF
FOR PROGRAM MOLCONN2
7 7 0 0 0
0.7943 -0.2132 0.0000 C 0 0 0 0 0
0.0023 -1.5022 0.0000 C 0 0 0 0 0
-1.5284 -1.4655 0.0000 C 0 0 0 0 0
-2.2648 -0.1072 0.0000 C 0 0 0 0 0
-1.4690 1.1987 0.0000 C 0 0 0 0 0
0.0565 1.1609 0.0000 C 0 0 0 0 0
2.3413 -0.2625 0.0000 O 0 0 0 0 0
1 2 2 0 0 0
2 3 1 0 0 0
3 4 2 0 0 0
4 5 1 0 0 0
5 6 2 0 0 0
6 1 1 0 0 0
1 7 1 0 0 0
M END
> 25 <BOILING POINT>
182.0
> 25 <MELTING POINT>
40.0 - 42.0
> 25 <ALTERNATE NAME>
Hydroxybenzene
> 25 <DATE>
10-02-92
$$$$
2-Chloro-4-nitro PHENOL
XXMACCS 8302248414282D 1 0.00213 0.00000 0 JF
FOR PROGRAM MOLCONN2
11 11 0 0 0
0.7943 -0.2132 0.0000 C 0 0 0 0 0
0.0023 -1.5022 0.0000 C 0 0 0 0 0
-1.5284 -1.4655 0.0000 C 0 0 0 0 0
-2.2648 -0.1072 0.0000 C 0 0 0 0 0
-1.4690 1.1987 0.0000 C 0 0 0 0 0
0.0565 1.1609 0.0000 C 0 0 0 0 0
2.3413 -0.2625 0.0000 O 0 0 0 0 0
1.0 1.0 0.0000 Cl 0 0 0 0 0
2.0 2.0 0.0000 N 0 0 0 0 0
3.0 3.0 0.0000 O 0 0 0 0 0
4.0 4.0 0.0000 O 0 0 0 0 0
1 2 2 0 0 0
2 3 1 0 0 0
3 4 2 0 0 0
4 5 1 0 0 0
5 6 2 0 0 0
6 1 1 0 0 0
1 7 1 0 0 0
2 8 1 0 0 0
4 9 1 0 0 0
9 10 2 0 0 0
9 11 2 0 0 0
M END
> 25 <MELTING POINT>
85.0 - 87.0
> 25 <PHYSIOLOGICAL>
IRRITANT
$$$$
(Note "blank" record to terminate file!!!)
The test input file SDF.B is supplied with the Molconn-Z software.
Molfile format is licensed by MDL Information Systems, Inc.
San Leandro, CA
SMILES Strings using the Daylight Toolkit Format (unix only)
The SGI and (potentially) some other UNIX versions of standalone Molconn-Z have the added
capability of reading and decoding SMILES files using the Daylight Toolkit SMILES interpreter
instead of the built-in Molconn-Z SMILES interpreter. This is an
optional feature that requires additional licensing from eduSoft, LC/Hall Associates and a
run-time SMILES Toolkit license from Daylight Chemical
Information Systems, Inc. Each record of the Daylight SMILES files, which are
generally named with the .smi extension, is simply a SMILES String followed by the
Molecule Name (space delimited). There is no file termination code. This file format matches what is
supported by the Daylight database software and be a useful option for some sites
that have large databases already encoded in this way. The other potential advantage is that
the Daylight Toolkit is the defacto standard for interpretation
of SMILES codes; while we have done no head-to-head comparisons and can cite no
specific examples, we believe that the Daylight methodology is probably more robust
for this function than the built-in Molconn-Z decoder and would recommend, for those
who plan to work with SMILES on unix computers, to consider licensing this option.
The File smallmol.smi as Supplied in Unix Versions of Molconn-Z Software:
c1ccccc1 benzene
C(Cl)(Cl)Cl chloroform
CC ethane
C1CCCCC1 cyclohexane
CC(C)(C)O tbutanol
c1cccc2ccccc12 napthalene
C1(O)C(O)C(O)C(CO)OC1OC(C(CO)O)C(O)C(O)C(=O)O maltobionic_acid
c1ccccc1CC(N)C amphetamine
c1cc(C)ccc1Cc(cc2)ccc2C di_p_tolyl_methane
Molecule Files from User Supplied File Formats
Molconn-Z has several options for use of input file format. The input option is selected as item 1 in
the MENU section of the program. This option is selected as 'U' in option 1 in the Main Menu section
of the program.
The Molconn-Z program is currently set up to accept input files in several formats. If the user has a
different file format for molecules, the user may request the object code for Molconn-Z with an appropriate
entry point subroutine (USERFIL) that can be end-user coded to accept these files as input. Alternatively,
the user may contact Hall Associates for the possibility of a customized version of USRFIL.
The SAMPLE.B Example File
Several example input .B files are supplied with the Molconn-Z software. These files have been described
in the above sections. The section on the standard MOLCONN format listed three example molecules. Below
is given the contents of the example file for standard MOLCONN format called SAMPLE.B. The user may
find this useful in learning how to use Molconn-Z with the standard MOLCONN format.
The File SAMPLE.B as Supplied in Molconn-Z Software:
1, Propanol
1,3,C,2
2,2,C,1,3
3,2,C,2,4
4,1,O,3
-1
2, 2-Propanol
1,3,C,2
2,1,C,1,3,4
3,1,O,2
4,3,C,2
-1
3, Aniline
1,0,C,2,6,7
2,1,C,1,3
3,1,C,2,4
4,1,C,3,5
5,1,C,4,6
6,1,C,5,1
7,2,N,1
-1
4, Benzyl alcohol
1,0,C,2,6,7
2,1,C,1,3
3,1,C,2,4
4,1,C,3,5
5,1,C,4,6
6,1,C,5,1
7,2,C,1,8
8,1,O,7
-1
5, 3-Bromo phenol
1,0,C,2,6,7
2,1,C,1,3
3,0,C,2,4,8
4,1,C,3,5
5,1,C,4,6
6,1,C,1,5
7,1,O,1
8,0,Br,3
-1
6, Benzimidazole
1,1,N,2,5
2,1,C,1,3
3,0,N,2,4
4,0,C,3,5,9
5,0,C,4,1,6
6,1,C,5,7
7,1,C,6,8
8,1,C,7,9
9,1,C,8,4
-1
7, Adamantyl amine
1,0,C,2,8,9,11
2,2,C,1,3
3,1,C,2,4,10
4,2,C,3,5
5,1,C,4,6,9
6,2,C,5,7
7,1,C,6,8,10
8,2,C,1,7
9,2,C,1,5
10,2,C,3,7
11,2,N,1
-1
8, Ephedrine
1,0,C,2,6,7
2,1,C,1,3
3,1,C,2,4
4,1,C,3,5
5,1,C,4,6
6,1,C,5,1
7,1,C,1,8,9
8,1,O,7
9,1,C,7,10,11
10,3,C,9
11,1,N,9,12
12,3,C,11
-1
9, 4,4'-Dichloro biphenyl
1,0,C,2,6,7
2,1,C,1,3
3,1,C,2,4
4,0,C,3,5,8
5,1,C,4,6
6,1,C,5,1
7,0,Cl,1
8,0,C,4,9,13
9,1,C,8,10
10,1,C,9,11
11,0,C,10,12,14
12,1,C,11,13
13,1,C,8,12
14,0,Cl,11
-1
-1