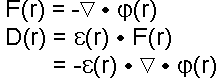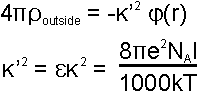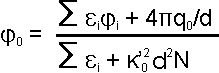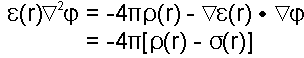:
:
Zap is a new program for solution of the Poisson-Boltzmann Equation (also referred to as the PBE). This is an approach that allows the calculation of a variety of electrostatic properties for neutral and charged molecules. Electrostatic interactions are very important to many biological processes. To understand the nature of these interactions it is crucial to characterize the physical and chemical properties of these biological molecules in solution, which is their natural state. While the principles of solvent effects on solutes have been known for a long time, the application of these principles to enable predictions of experimentally-observable properties have not been available. Recently, with the advent of structure-based drug design and protein engineering, and the growing interest in de novo predictions of protein folding, the importance of accurately describing the electrostatic environment of biomacromolecules has come to the forefront.
The most accurate model for understanding the properties of aqueous solutions would be to explicitly model all molecules in the system, including the solvent molecules using a molecular mechanics approach. The electrostatically important portions of a typical molecular mechanics force field are the Coulombic and van der Waals terms. Of course this would entail calculations of interactions between each solvent molecule and each "solute" molecule as well as between the solvent molecules themselves. Also significant is that this calculation would have to be performed and averaged for many solvent molecule configurations to account for molecular motion and entropy. While this is possible for some systems on a conventional workstation, and other systems on a supercomputer, it is not particularly practical.
A complementary approach, using the empirical free energy information from LogPo/w, which implicitly incorporates solvation effects, is provided by the HINT program. HINT is also available from eduSoft LC.
Another alternative is to use continuum or macroscopic methods, which treat solvents not as discrete molecules, but as environmental properties with average values. In particular, the electrostatic properties of solvents such as water can be described with descriptors for charge, dielectric, etc. In this chapter we will discuss the solution of the PBE as implemented in programs such as DelPhi, followed by a description of the enhancements that are incorporated in the Zap algorithms.
In classical electrostatics materials are
considered to be homogeneous dielectric media that can be polarized
by electrical charges. A dielectric constant can be considered a bulk
measure of the polarizability of the media. This is a
continuum model because the polarization of each atom is not
considered. Coulomb's law, eqn. 1, describes the Electrostatic
energy, Eij in terms of two charges, qi and qj,
the distance between the atoms, rij and the dielectric
constant
:
|
|
(eqn. 1) |
The potential,
 i
at atom i is given by:
i
at atom i is given by:
|
|
(eqn. 2) |
For instance, two charges of +1 separated
by one Ångstrom in vacuum (,
dielectric = 1.0) have an energy of 560.9 kT (where k is Boltzmann's
constant and T is temperature in K) or 331 kcal/mol. It is important
to note that this model is only valid for an infinite medium of
uniform dielectric, which is certainly not the actual case in a
complex solution of biomacromolecules, ligands and polar solvent
(like water). And of course, these are the types of problems we are
most interested in.
Biomacromolecules such as proteins have rather low dielectrics (typically 2-5) because their polar groups and side chains are more or less frozen in place and cannot actually reorient significantly in response to an externally applied dielectric field. Also, many portions of macromolecules are apolar. However, water has a dielectric constant of around 80 and is able to re-orient its dipoles freely. Thus a biomacromolecular system in aqueous solution has two extremely different dielectric properties for the two components, macromolecule and water. The Poisson-Boltzmann Equation (PBE) is capable of modeling this type of situation.
The electrostatic field, F, and electrostatic displacement, D, as functions of r can be written as:
|
|
(eqn. 3) |
Note that
is rigorously a tensor, i.e. the polarization can, in theory, be in a
different direction from the polarizing field. In practice this is
ignored and it is treated as a scalar quantity. In an inhomogeneous
dielectric media with charges, Poisson's equation relates the
electric displacement to the charge density
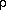 (r):
(r):
|
|
(eqn. 4) |
By substituting D(r) from eqn. 3,
|
|
(eqn. 5) |
It is useful to separate the charge
density,
(r),
into two parts:
|
|
(eqn. 6) |
where
inside
is the charge distributions of all the fixed charges inside
the molecule, and
outside
is the charge density outside the molecule (i.e., solvent)
which accounts for reorientation and redistribution of (ion) charges
in the solution.
The charge
density,
outside,
can be modeled by a Boltzmann distribution. If the charge density of
the molecule is low then:
|
|
(eqn. 7) |
where
is the dielectric constant of the solvent,
is the electrostatic potential in units of kT/e, e is the charge of
an electron, NA is Avogadro's number, I is the ionic
strength in moles/liter, k is Boltzmann's constant, T is temperature
in K, and
 '
is the Debye-Hückel inverse length. Note that
'
appears with a negative sign because charge accumulates of opposite
sign to the inducing potential.
'
is the Debye-Hückel inverse length. Note that
'
appears with a negative sign because charge accumulates of opposite
sign to the inducing potential.
Substituting eqn. 7 into the Poisson equation (eqn. 5) gives the linear form of the Poisson-Boltzmann equation:
|
|
(eqn. 8) |
where
'0
is 0 inside the molecule and
'
outside the molecule (i.e., in the solvent).
Because it includes the effects of solvent/molecule dielectric as well as ionic strength, the Poisson-Boltzmann equation is a good model for the electrostatic properties of complex biological systems, except that it cannot be solved analytically for any but the simplest dielectric boundary shapes; e.g., spheres, etc. Numerical approximations such as the Finite Difference Algorithm are required to solve the PBE for the complex shapes and structures of biological molecules in solution.
The numerical solution usually chosen to
solve the PBE is to map the molecule onto a three dimensional grid
and apply the finite difference approximation. A two dimensional
representation of this is shown below. Note that the grid spacing is
shown as d, and that the interior of the molecule is assigned a
dielectric
inside
different from the exterior of the molecule (solvent) which is
assigned a dielectric
outside.
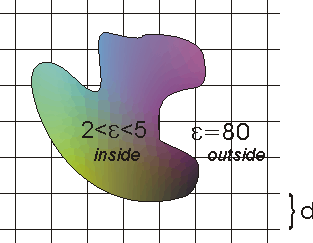
The PBE must be satisfied everywhere in the
system, but most importantly at each of the grid points. If a cube of
dimension d x d x d is considered to surround each grid point, the
derivatives in eqn. 8 can be replaced by "finite differences"
measured in the cube. Thus, the functions
,
and
can be represented by their values at grid points.
In the figure above, the black sphere
represents the grid point of interest (i=0), the blue spheres are the
six neighboring grid points (i=1-6). For each of these seven points
in space are associated values of qi (charge),
i
and
'0.
The yellow spots, on the surface of the cube surrounding grid point
0, identify the dielectric constants, which are calculated half-way
between grid point 0 and each of its six neighbors.
The potential at any grid point (0) depends
on q0,
'0
with grid spacing, d, and the potential,
i,
and dielectric,
i,
of the neighboring six grid points:
|
|
(eqn. 9) |
where N = 1 for the linear case (eqn. 8) or
N = (1 +
02/3!
+
04/5!
+ ...) for the non-linear case if the charge density of the molecule
is not low and/or the ionic strength of the solution is not low.
From this the potential at each grid point can be calculated. It is important to note that these potentials will adjust as a function of the potential of the neighboring grid points. Thus, this calculation is iteratively repeated to increase the accuracy and precision of the potentials. A set of convergence criteria must be established for each run.
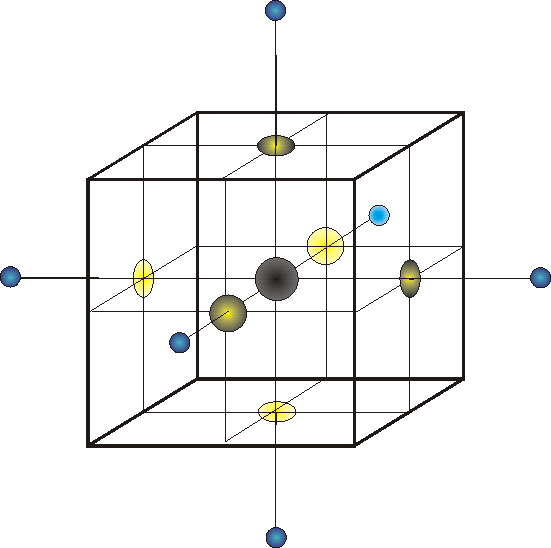
The most basic electrostatic quantity is the electrostatic potential. The potential at a charge can be considered to be comprised of three terms: the Coulombic potential from all the other charges (figure a), the reaction of the solvent to the charge (figure b), the reaction of the solvent to all the other charges (figure c).
|
|
(eqn. 10) |
The utility of this decomposition is that it represents: The regular vacuum force-field (a);
the "self-energy" of the charge in the context of the molecule's dielectric (b);
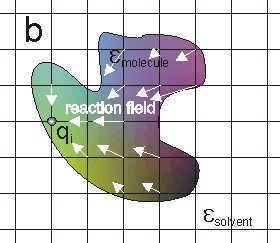
and the "screening" interaction of the solvent on charges within the molecule (c).

This, when added to the potential at a
charge due to its own field,
i(i),
yields the total potential.
|
|
(eqn. 11) |
Note that while the potential at a charge due to its own Coulomb field is infinite, it is easy to remove analytically (simply not including it in the sum over charges). It appears on the grid as a non-infinite, artifactual, potential. This can be removed by analytic or numerical techniques.
What makes ZAP different from previous PBE solvers? Consider the PBE (eqn. 5) rewritten so:
|
|
(eqn. 12) |
The second term on the LHS (left hand side) is the Laplacian operator. If this were the only LHS term the solution would be Coulomb's Law (eqn. 1). The first term represents the polarization of the solvent and is non-zero only where the dielectric constant is varying.
As is shown in eqn. 13, this can be thought
of as a polarization "surface" charge,
 (r).
Some methods of solving the PBE explicitly make use of this
reformulation: Boundary Element methods explicitly calculate the
polarization charge and then use this to calculate potentials within
or exterior to the molecule.
(r).
Some methods of solving the PBE explicitly make use of this
reformulation: Boundary Element methods explicitly calculate the
polarization charge and then use this to calculate potentials within
or exterior to the molecule.
|
|
(eqn. 13) |
The problems of grid-based methods such as DelPhi arise because the physical description of the dielectric is discontinuous. It is low inside the molecule, high outside and the transition between is infinitely sharp. Not only is this unphysical, it is very hard for grids to represent because the transition is unlikely to fall exactly on a grid point.
ZAP solves this problem by basing the dielectric description upon atomic-centered gaussians. This creates a dielectric function that varies from internal to external values smoothly over a range of a few 1/10th of an Ångstrom. The ZAP solution of the PBE is therefore much more robust and precise. (In addition, because we can easily calculate the derivative of the dielectric, a simple numerical trick allows us to calculate solvent forces.)
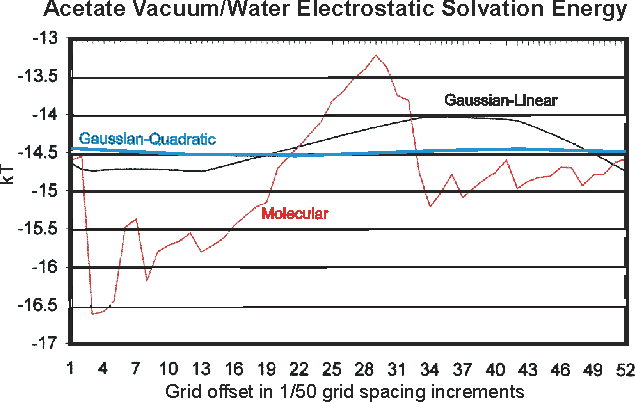
An example of the improved behavior of ZAP over DelPhi is shown in the figure below. Here we have changed the "register", i.e. the precise placement of the molecule on the grid, by successive increments of 0.02 of a grid spacing and plotted the energy due to the solvent reaction field. The DelPhi-like approach (labeled "Molecular" on the graph) leads to a jagged, almost random-walk line. The two ZAP curves represent different ways of mapping the molecular charge on to the grid. The older, DelPhi-like method leads to a curve that still retains some kinks and has substantial variation. When the new ZAP method (quadratic interpolation) is used the variation becomes smooth and almost negligible.
As a grid-based PBE solver, ZAP naturally produces grid output. The most basic of these is the raw potential map. Each grid point is assigned a potential (in kT/e), i.e. a value of 1.0 means the energy for a unit charge at this point is 1 kT (0.591 kcal/mol). The potentials inside the molecule from this map are not particularly interesting because they are dominated by the atomic charges. Outside the molecule, however, potentials take on the character of the solvent-screened field.
Another map that ZAP can produce is the effective induced charge from the solvent, i.e. the first term in eqn. 12, which represents the net solvent polarization. For PBE models where the dielectric varies discontinuously at the surface, this charge forms an infinitely thin layer around the molecule. In ZAP the surface charge is distributed around the surface in a layer of a few tenths of an Ångstrom.
Quantities that can be calculated from the
raw potential map are the atom potentials and the atom-by-atom
contributions to the solvation energy. The first have a slight
complication: the actual potential at a charge is infinite due to its
own Coulomb field,
i(i).
This is overcome by analytically removing this self-contribution. The
result is the potential at each atom from the solvent and every other
atom in the molecule. The second set of quantities is the potential
produced by the solvent at each atom multiplied by half the charge on
that atom. The sum of this quantity over all atoms is the energy of
bringing a molecule from a media of dielectric equal to the molecule
(e.g. organic solvent) into water.
Other electrostatic and molecular properties may be derived from the ZAP calculation. These include:
Total Molecular Charge
Sum over Absolute Value of Charges
Surface Area (Molecular Surface or Accessible Surface)
Fraction of Surface Area between specified or default ranges
Number of patches of surface greater than or less than specified or default thresholds
Fractional area of largest patch greater than or less than specified or default thresholds
Molecular Volume
Molecular Steric Quadrapole Moments (three)
Contracted Steric Octapole Moments (three)
The first two quantities are straightforward. The net charge is obvious and the sum of the absolute value of each atomic charge is a measure of molecular polarity (e.g. polar molecules will have a larger value than non-polar).
Surface area is a key concept in many molecular descriptors and ZAP provides either the accessible (that formed by expanding the radius of each atom by the radius of water ~ 1.4 Å) or the molecular (that formed by the inner surface of a ball the radius of water rolled over the molecule) area. Each point on a surface can be assigned a potential from the raw ZAP potential map. We can contract this information in several ways. Given certain potential thresholds we can ascertain what fraction of a surface falls between or beyond these limits. For example, limits of -1 and +1 kT would produce three fractions: surface of potential less than -1 kT, surface of potential between -1 and 1 kT, and surface of potential greater than +1 kT. ZAP contains default threshold values that have been designed to produce contributions to each range or "bin" for typical molecules.
Another analysis of surface information is via connectivity. If we remove all parts of the surface that are less than a given potential the resultant surface will consist of "islands" of positive potential. For simplicity ZAP only reports the number of such and the size of the largest as a fraction of the total surface area. We may also wish to set a negative threshold, i.e. remove all surface with potential greater than some value to calculate negative patches. These thresholds may be set by the user or left to default to the maximum and minimum threshold values from the range analysis described above.
Finally, there are volume characteristics. This has relevance to electrostatics as it represents the amount of physical space assigned a low dielectric value. The first such parameter is the molecular volume. The second are a triplet that are analogous the the moments of inertia, and the third are a triplet that measure the degree of deviation from ellipsoidal symmetry.
Honig, B.; Nicholls, A. "Classical Electrostatics in Biology and Chemistry". Science 1995, 268, 1144-1149.
Klapper, I.; Hagstrom, R.; Fine, R.; Sharp, K.; Honig, B. "Focussing of Electric Fields in the Active Site of Cu-Zn Superoxide Dismutase: Effects of Ionic Strength and Amino Acid Modification". Proteins Str. Funct. Genet. 1986, 1, 47-59.
Gilson, M.K.; Sharp, K.; Honig, B. "Calculating the Electrostatic Potential of Molecules in Solution: Method and Error Assessment". J. Comp. Chem. 1987, 9, 327-335.
Gilson, M.K.; Honig, B. "Calculation of the Total Electrostatic Energy of a Macromolecular System: Solvation Energies, Binding Energies and Conformational Analysis". Proteins Str. Funct. Genet. 1988, 4, 7-18.
Nicholls, A.; Honig, B. "A Rapid Finite Difference Algorithm, Utilizing Successive Over-Relaxation to Solve the Poisson-Boltzmann Equation". J. Comp. Chem. 1991, 12, 435-445.
Yang, A.-S.; Gunner, M.R.; Sampogna, R.; Sharp, K.; Honig, B. "On the Calculation of pKas in Proteins". Proteins Str. Funct. Genet. 1993, 15, 252-65.